Influenza A virus (IAV) is responsible for 3-5 million severe cases every year, resulting in 250 to 500 000 deaths. Most influenza strains evolve exclusively in water birds, but some highly pathogenic avian strains (
e.g. H5N1, H5N8, H7N9) can infect humans with lethal consequences (up to 60% mortality) and are potential pandemic threats for humanity if they develop human-to-human transmissability. In order for these avian viruses to efficiently replicate in mammalian cells, host adaptation of the viral polymerase is necessary. However, nothing is known about the molecular basis of this adaptation.
It has been known for many years that these adaptive mutants are localised on the C-terminal (627-NLS) domains of the PB2 subunit of the viral polymerase. In particular, mutation of PB2 residue 627 from E to K in avian polymerase. It has also been shown that A host
transcription regulator ANP32A, comprising a long C-terminal
intrinsically disordered domain (IDD), has also been shown to be responsible for this viral adaptation. Intriguingly, human ANP32A IDD lacks a 33 residue insertion compared to avian ANP32A, a deletion that restricts avian influenza polymerase activity in mammalian cells. Nothing however was known about the molecular basis of this adaptation, probably because both the 627-NLS region and the host protein were highly dynamic, involving long intrinsically disordered domains that do not form unique structures or crystallise.
Using NMR spectroscopy, researchers at the IBS determined conformational ensembles of the highly dynamic complexes between E627 and K627 forms of the 627-NLS domains of PB2 and avian and human ANP32A. They reveal that the negatively charged IDD of human ANP32A transiently and preferentially binds to a basic face of the 627 domain, exploiting multiple binding sites to maximize affinity for 627-NLS. The presence of E627 interrupts the polyvalency of the interaction, an effect that is compensated by extending the interacting regions and exploiting an avian-unique motif in the unfolded domain of ANP32A that interacts with the 627-NLS linker (Figure). Importantly the two binding modes exploited by human- and avian-adapted PB2 to adapt to the differences between the avian and host proteins are impossible in the cross interaction between avian polymerase and human ANP32A, suggesting that this molecular specificity may be related to species adaptation. In collaboration with the laboratory of Stephen Cusack at the EMBL Grenoble they could also show that the observed binding mode is maintained in the context of heterotrimeric influenza polymerase, placing ANP32A in the immediate vicinity of known host-adaptive PB2 mutants.
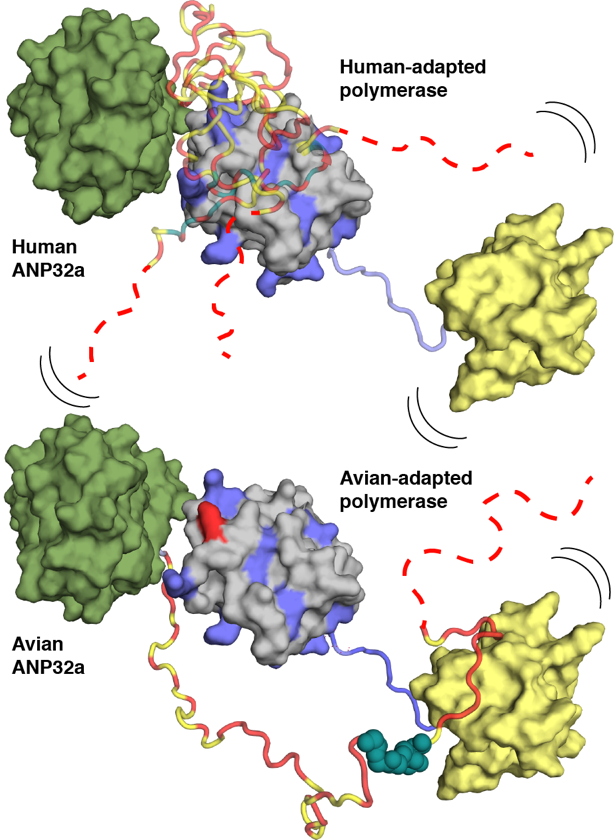
This study provides a molecular framework for understanding the differential binding modes underlying species-specific restriction of influenza polymerase by ANP32A and will inform the identification of new targets for influenza inhibition.
Transcription regulation is the phase of controlling the expression of genes acting at the level of DNA transcription. In this study, these are ANP32A for the avian form and ANP32A IDD for the human form.
Intrinsically disordered proteins lack a stable three-dimensional structure and are functional in their disordered state. Their high flexibility allows them to adapt easily to the surface of their partners. They are thus able to fold up during interaction.