Metastatic kidney cancer is a highly vascularized cancer. Over the last decade, targeted therapies have been developed including multi-target tyrosine kinase inhibitors (sunitinib, sorafenib, axitinib and pazopanib), serine-threonine kinase inhibitors such as temsirolimus, everolimus. Since its approval in 2006, sunitinib is the first-line care standard for metastatic renal cancer with significant improvement in survival. However, resistance to treatment may occur after 6 to 36 months. In order to predict this resistance very early, there is a need of biomarkers.
In 2013, the scientists from the team «Invasion Mechanisms in Angiogenesis and Cancer» of the Biology of Cancer and Infection Laboratory have identified a soluble form of
VE-cadherin* in blood from glioblastoma patients
[1]. In order to understand how a transmembrane protein exclusively expressed in endothelial cells (EC) was released in blood, the researchers performed cell experiments with the angiogenic growth factor VEGF, the major actor involved in tumor vascularization. They showed that VEGF induced the tyrosine phosphorylation of VE-cadherin in its cytoplasmic domain
via the activation of the tyrosine kinase Src. They also demonstrated that this process preceded the release of the extracellular domain of the protein (sVE) (Figure). These post-translational modifications are responsible for EC destabilization which is a characteristic of EC from tumor capillaries.
Then the scientists aimed at determining whether antiangiogenic treatment commonly used for the clinical care of metastatic kidney cancer had an effect on VE-cadherin post-translational modifications (i.e., phosphorylation and cleavage) and if so, whether such modifications were helpful to monitor patient outcome. The results show that
[2], on a EC monolayer model mimicking the vascular endothelium, sunitinib blocks the early effects of VEGF on VE-cadherin which was not the case of serine-threonine kinase inhibitors or other therapies. In a 3 D culture model (spheroids) with EC and tumor cells, sunitinib strongly inhibited EC migration. These data suggest that Sunitinib has an impact on VE-cadherin in maintaining its structure to restore EC integrity.
A strong collaboration with clinicians has allowed to analyse sVE in blood from metastatic kidney cancer patients (n=131) before and after 4 weeks of Sunitinib treatment. sVE was detected at baseline which level was higher than in a healthy donor group (n = 96). Analysis of sVE level after 4 weeks of treatment showed that a decrease in sVE level discriminates the responders vs. non-responders to sunitinib. No decrease is observed in non-responder patients. The mechanism of this nonresponse remains to be studied. These data highlight the interest for the sVE bioassay in future follow-up of cancer patients treated with targeted therapies such as tyrosine-kinase inhibitors in the field of personalized medicine.
These works, of which H. Polena, and A Khalil-Mgharbel post-doctoral positions, were financed by ARC Foundation and LIGUE contre le Cancer (committee Isère).
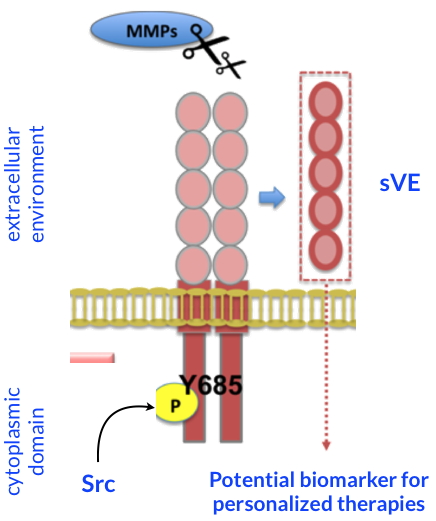
Post-translational modifications of VE-cadherin.
VEGF induces structural modifications of VE-cadherin (Y685 phosphorylation and cleavage of its extracellular domain) upon Src pathway. Cleavage of the extracellular domain of VE-cadherin is dependent on extracellular proteases (MMPs) secreted by the tumor cells. These changes are blocked by the sunitinib. These data propose sVE as a potential candidate biomarker for personalized therapies.
*
VE-cadherin is a specific biomarker of endothelial cells whose phosphorylation and cleavage contribute to the destabilization of endothelial junctions.